This page gives pointers to software written and maintained by other groups which can be used with LAMMPS. The first category is external LAMMPS packages which can be downloaded and compiled with LAMMPS to extend its capabilities. The second category is stand-alone tools which either perform calculations such as force-field fitting that generate complex input for LAMMPS, or that use LAMMPS as an MD engine.
We think these kinds of tools, whether freeware or commercial, can be very useful. They extend the scope of problems that LAMMPS can model. We are happy to advertise software here, so that LAMMPS users can try it out. Send us an email if you want to add something to the list.
NOTE: ML = machine learning
URL: https://github.com/atzberg/mlmod
Author: Paul Atzberger (University of California Santa Barbara), (atzberg at gmail.com)
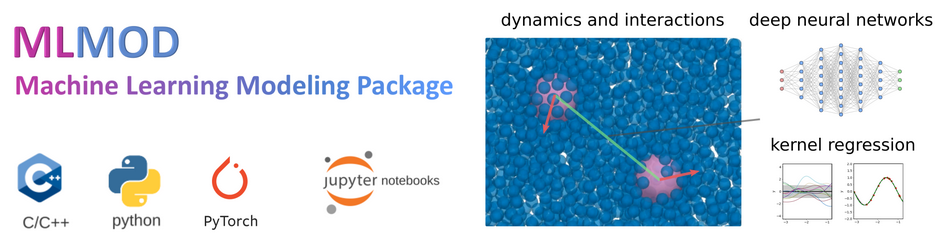
MLMOD-PYTORCH is a Python/C++ package for utilizing machine learning methods and data-driven modeling for simulations in LAMMPS. The package provides methods for time-step integrators for dynamics and interactions using general ML model classes, including Neural Networks, Kernel Regression, and others. Models can be trained and exported from PyTorch or from other machine learning frameworks.
A complete list of features, downloads, and tutorials can be found at the URL.
MLMOD-PYTORCH is organized as a stand-alone library libmlmod.so with a lightweight interface to LAMMPS via C++ code in a patch folder USER-MLMOD.
For a quick start installing and using the package, see the URL for the python pip install command with pre-compiled binaries, or links to avaialbe docker images.
More details on the package and aims for future releases can be found in the following paper: https://arxiv.org/abs/2107.14362
Author: Paul Atzberger (University of California Santa Barbara), (atzberg at gmail.com)
SELM stands for Stochastic Eulerian Lagrangian Method. This is a set of numerical methods that utilizes a mixed Eulerian description for hydrodynamic fields coupled to a Lagrangian description of the coarse-grained degrees of freedom. To account for both hydrodynamic flow and thermal fluctuations, fluctuating hydrodynamic equations are introduced and coupled to the coarse-grained degrees of freedom in a manner consistent with statistical mechanics. For implicit-solvent coarse-grained models, the methods can be used to account for momentum transfer through the missing solvent degrees of freedom. The methods provide alternative thermostats to the widely used Langevin thermostat and Dissipative-Particle-Dynamics thermostat.
SELM is provided as a USER-SELM package for LAMMPS, which implements several fluctuating hydrodynamics thermostats for different physical regimes:
A complete list of features, downloads, and tutorials can be found on the package homepage: http://mango-selm.org.
Please feel free to register and download MANGO-SELM from our website. For questions or suggestions, please use the listed MANGO-SELM discussion forums.
URL:https://github.com/HidekiMori-CIT/aenet-lammps
Author: Hideki Mori, College of Industrial Technology, Japan (morih.cit.01 at gmail.com)
Using this package, LAMMPS users can perform MD and MM simulations with ANN atomic potentials generated by aenet; see http://ann.atomistic.net for more details. This package allows for flexible conditions by adopting conventional pair_style and pair_coeff formats like EAM.
With the plugin command it is possible to load additional LAMMPS styles into an LAMMPS executable at runtime, if compiled accordingly. The lammps-plugins repository contains source code for several external LAMMPS styles updated for recent versions of LAMMPS and combined with a plugin loaded and a CMake build system to compile them into plugins. Please see the README file on the GitHub repo for the list of included packages and styles.
Please note that the USER-DEEPMD package can also be configured and compiled as a plugin to extend a compatible, existing LAMMPS executable.
For more information about LAMMPS plugins, please see the manual
URL: https://github.com/sntioudis/papreca
Author (developer and methodology): Stavros Ntioudis (Imperial College London) (stavros.ntioudis20 at imperial.ac.uk)
Contributors (methodology): James P. Ewen (University of Bath), Daniele Dini (Imperial College London), C. Heath Turner (University of Alabama)
PArallel PREdefined CAtalog (PAPRECA) is a hybrid off-lattice kinetic Monte Carlo/molecular dynamics (kMC/MD) framework. The software offers a scalable avenue for kMC/MD calculations. It couples a parallel off-lattice kMC solver with LAMMPS (used as an MD engine library) to resolve both high-activation-energy processes (e.g., adsorption) and high-frequency events (e.g., atomic vibrations). The framework is flexible and supports a variety of predefined off-lattice events such as reactions, adsorption/desorption steps, and diffusion hops. PAPRECA is developed in C++, but similarly to LAMMPS, simulations are defined through input files, enabling use without prior C++ experience. Example applications include but are not limited to adsorption/desorption on catalytic surfaces, amorphous thin films, and modeling self-diffusion of gases or solids.
Information about the off-lattice kMC/MD framework can be found in our relevant articles:
1 Ntioudis et al. (2024), Journal of Open Source Software, 9(98), 6714. https://doi.org/10.21105/joss.06714
2 Ntioudis et al. (2023), Computational Materials Science, 229, 112421. https://doi.org/10.1016/j.commatsci.2023.112421
A complete list of features, downloads, and tutorials can be found on the package homepage: https://sntioudis.github.io/papreca/
URL: https://github.com/orsim/elba-lammps
Author: Mario Orsi (orsimario at gmail.com)
ELBA-LAMMPS is a toolkit that assists LAMMPS users in simulating the ELBA coarse-grain model, as described in Orsi & Essex, PLoS One 6: e28637 (2011) and illustrated in the following figure. The ELBA-LAMMPS package includes specific installation guidelines, simulation examples, analysis scripts, and tools to enable visualization with VMD.
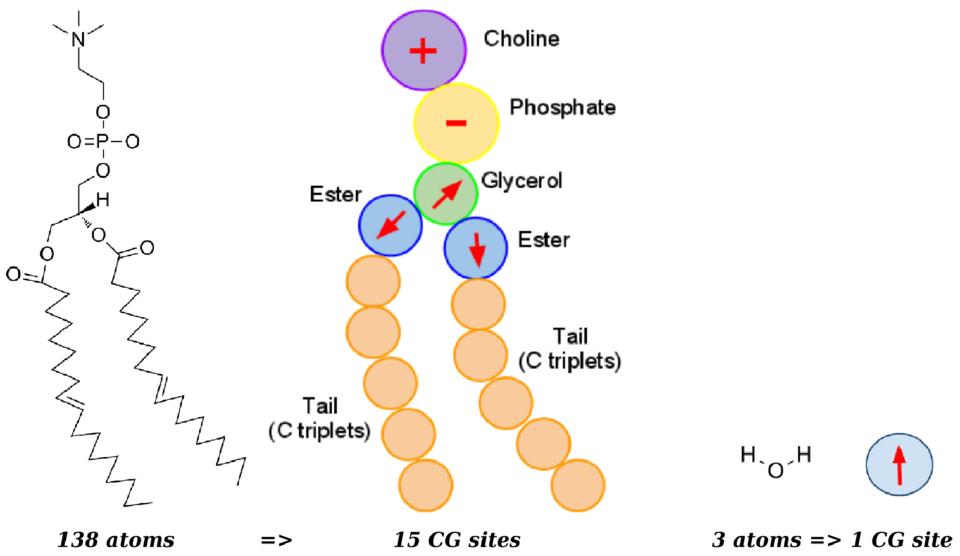
ELBA coarse-grain model of a DOPC phospholipid (left) and a water molecule (right). Coarse-grain electrostatics are represented by positive ("+" sign) and negative ("-" sign) point charges, and point dipoles (arrows). Click on image to view larger version.
URL: https://github.com/patherlkd/easy-pair-table-lammps
Author: Luke K. Davis (luke.davis at ucl.ac.uk)
EPSL (easy-pair-table-lammps) is a python tool to create any pair potential to use with LAMMPS' pair_style table, and provides an automatic check against the pair_write output.
Author: Christoph Kloss (JKU)
LIGGGHTS stands for LAMMPS Improved for General Granular and Granular Heat Transfer Simulations. It is a stand-alone code focused on granular material modeling, which uses LAMMPS intnerally as a particle engine. It implements the following capabilities on top of the LAMMPS "GRANULAR" features:
More features, such as improved handling for non-spherical particle, a 6 degrees of freedom solver for arbitrarily shaped bodies and wall stress analysis, are currently under development. Also, an efficient parallel coupling to the OpenFOAM(R) framework is under development.
LIGGGHTS will stay "backward compatible" to LAMMPS, meaning one can use all powerful LAMMPS features.
For a complete list of features, videos, as well as downloads and tutorials, please refer to the URLs above.
If you are interested in learning more about DEM with LIGGGHTS, there will be 2 classes (each for 3 days) held in the city of Linz, Austria. They are scheduled for June 9-11 and September 22-24. If you are interested in joining one of the classes, please answer to this email.
Feel free to register for free and download LIGGGHTS 1.0 from our website. For any question that may arise, please use the provided discussions forums.
Authors: see PLUMED WWW site
PLUMED is a plugin for free-energy calculations in molecular systems that can be interfaced with some of the most popular molecular dynamics codes using a simple patching procedure.
PLUMED allows one to perform several type of calculations, including:
It has the following features:
Authors: see VOTCA WWW site
VOTCA is a set of tools for creating corase-grained models, as well as simulating charge transport, and has other features as well.
Here is a summary of the methods in the coarse-graining toolkit (VOTCA-CSG):
It has support for processing LAMMPS dump files.
URL: http://www.wag.caltech.edu/home/ajaramil/GARFfield.html
Author: Andres Jaramillo-Botero (Caltech, ajaramil at caltech.edu or ajaramil at gmail.com)
GARFfield is a multi-platform (I have tested it on my Mac OS X and on multiple Linux distributions), multi-objective parallel hybrid genetic algorithm/conjugate-gradient based force field optimization framework. It enables first-principles based force field parameter optimization from quantum mechanical and phenomenological data sets. In principle, the latter allows improved accuracy and transferability over a wider range of molecular compositions, interactions and environmental conditions unexplored by experiments. GARFfield currently supports multiple reactive and non-reactive force fields through LAMMPS. It is particularly well suited to develop adiabatic and non-adiabatic reactive force fields, such as ReaxFF (REAX or USER-REAXC) and eFF-ECP (USER-EFF), respectively allowing relative restraints on bond-order based bonds, angles, torsions, transition states, and also on electron sizes. GARFfield can produce Pareto-optimal parameters, while allowing hill-climbing from local minima solutions, fixed or random weights on training sets, and deterministic conjugate-gradient minimization near a parabolic minima for improved convergence.
New features are currently being developed, including template-based force field support (to avoid syntax dependencies from LAMMPS), heuristic sequencing in parameter optimization, and training set parallelization (in addition to the currently available parallelization at the level of the genetic algorithm string population).
Please refer to the GARFfield User Manual (http://www.wag.caltech.edu/home/ajaramil/GARFFIELDUserManual.pdf) and to the paper by A. Jaramillo-Botero, S. Naserifar., and W.A. Goddard III, "A General Multi-Objective Force Field Optimization Framework, with Application to Reactive Force Fields for Silicon Carbide", Journal of Chemical Theory and Computation, 2014, 10 (4), pp 1426-1439. DOI: 10.1021/ct5001044 for additional information on capabilities and usage.
We are releasing the GARFfield code to promote a community-driven development effort, so If you want to contribute bug fixes, new extensions (e.g. force fields) or features (e.g. GUI) to GARFfield please register, download and compile GARFfield. We ask that once you integrate, validate and test your new extension or feature and submit it back to ajaramil at caltech.edu. Your contribution will then be reviewed and published as part of subsequent code releases.
Jazz is a physics research code used to calculate the lifetimes of vibrational modes in solids. The calculations are implemented as a Python wrapper for LAMMPS, which provides the energies and forces as calculated for any interatomic potential available in that package. The anharmonic character of the normal modes is calculated via the Monte Carlo-based moments approximation as is described in a series of papers by M. Daw and co-workers (D. Dickel, Y. Gao, and H. Wang). It is distributed as open-source software.
LEAD: Prof. Murray Daw (Clemson)
CONTRIBUTORS: Dr. Ted Dickel, Dr. Yang Gao, Mr. Hengjia Wang (were or are physics grad students at Clemson)
URL: https://jazzforlammps.sourceforge.net
URL: https://uspex-team.org/en/uspex/overview
USPEX is a method developed jointly by Artem R. Oganov, Andriy O. Lyakhov, Colin W. Glass and Qiang Zhu, and implemented in the same-name code written by Andriy O. Lyakhov, Colin W. Glass and Qiang Zhu. This method/code enables crystal structure prediction at arbitrary P-T conditions, given just the chemical composition of the material. Many previous attempts to solve crystal structure problem were plagued by low success rate and extreme computational costs that prevented full ab initio studies. USPEX avoids both of these problems. In fact, "uspekh" means "success" in Russian - which highlights a nearly 100% success rate that we find for our method.
USPEX can also be used for finding low-energy metastable phases, as well as stable structures of nanoparticles, surface reconstructions, molecular packings in organic crystals, and for searching for materials with desired physical (mechanical, electronic) properties. The USPEX code is based on an efficient evolutionary algorithm developed by A.R. Oganov's group, but also has options for using alternative methods (random sampling, metadynamics, corrected PSO algorithms). USPEX is interfaced with many DFT or classical codes, such as VASP, SIESTA, GULP, Quantum Espresso, CP2K, CASTEP, LAMMPS, and so on.
An extension of the Visual Studio Code editor for LAMMPS input files is available through the editor's embedded extension system. Alternatively, also through the vs-marketplace or GitHub.
The extension features:
See https://thfriedrich.github.io/lammps_vscode/ for more details